 Print Article
Print Article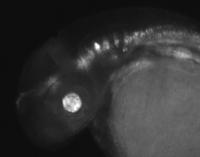
The glowing areas in this zebrafish embryo show the activity of one of the brain enhancer sequences identified. Johns Hopkins researchers have succeeded in teaching computers how to identify commonalities in DNA sequences known to regulate gene activity, and to then use those commonalities to predict other regulatory regions throughout the genome. The tool is expected to help scientists better understand disease risk and cell development.
The work was reported in two recent papers in Genome Research, published online on July 3 and Sept. 27.
"Our goal is to understand how regulatory information is encrypted and to learn which sequence variations contribute to medical risks," says Andrew McCallion, Ph.D., associate professor of molecular and comparative pathobiology in the McKusick-Nathans Institute of Genetic Medicine at Hopkins. "We give data to a computer and 'teach it' to distinguish between data that has no biological value versus data that has this or that biological value. It then establishes a set of rules, which allows it to look at new sets of data and apply what it learned. We're basically sending our computers to school."
These state-of-the-art "machine learning" techniques were developed by Michael Beer, Ph.D., assistant professor of biomedical engineering at the Johns Hopkins School of Medicine, and by Ivan Ovcharenko, Ph.D., at the National Center for Biotechnology Information. The researchers began both studies by creating "training sets" for their computers to "learn" from. These training sets were lists of DNA sequences taken from regions of the genome, called enhancers, that are known to increase the activity of particular genes in particular cells.
For the first of their studies, McCallion's team created a training set of enhancer sequences specific to a particular region of the brain by compiling a list of 211 published sequences that had been shown, by various studies in mice and zebrafish, to be active in the development or function of that part of the brain.
For a second study, the team generated a training set through experiments of their own. They began with a purified population of mouse melanocytes, which are the skin cells that produce the pigment melanin that gives color to skin and absorbs harmful UV rays from the sun. The researchers used a technique called ChIP-seq (pronounced "chip seek") to collect and sequence all of the pieces of DNA that were bound in those cells by special enhancer-binding proteins, generating a list of about 2,500 presumed melanocyte enhancer sequences.
Once the researchers had these two training sets for their computers, one specific to the brain and another to melanocytes, the computers were able to distinguish the features of the training sequences from the features of all other sequences in the genome, and create rules that defined one set from the other. Applying those rules to the whole genome, the computers were able to discover thousands of probable brain or melanocyte enhancer sequences that fit the features of the training sets.
In the brain study, the computers identified 40,000 probable brain enhancer sequences; for melanocytes, 7,500. Randomly testing a subset of each batch of sequences, the scientists found that more than 85 percent of the predicted enhancer sequences enhanced gene activity in the brain or in melanocytes, as expected, verifying the predictive power of their approach.
The researchers say that, in addition to identifying specific DNA sequences that control the genetic activity of a particular organ or cell type, these studies contribute to our understanding of enhancers in general and have validated an experimental approach that can be applied to many other biological questions as well. Source : Johns Hopkins Medicine
 Mail to a Friend
Mail to a Friend
