

Scientists from the Senckenberg and the Giraffe Conservation Foundation have analysed the genetic relationships of all major populations of giraffe in the wild. The large study on the genetic makeup of giraffe, published today in Current Biology, shows that there are four distinct giraffe species. Until now, only one giraffe species had been recognized. The unexpected results are based on analyses using several nuclear marker genes of more than 100 animals. The new insights are set to improve protection efforts of these endangered animals in Africa.
Category: Bioinformatics
| Bioinformatics | August 29, 2016 06:12 PM |
Researchers at Houston Methodist have developed an artificial intelligence (AI) software that reliably interprets mammograms, assisting doctors with a quick and accurate prediction of breast cancer risk. According to a new study published in Cancer (early online Aug. 29), the computer software intuitively translates patient charts into diagnostic information at 30 times human speed and with 99 percent accuracy.
| Full article | 0 Comments | 64537 views |
| Bioinformatics | August 23, 2016 06:09 PM |

This is an illustration of warbler. For decades, conservationists have considered blue-winged warblers to be a threat to golden-winged warblers, a species being considered for federal Endangered Species protection. Blue-winged warbler populations have declined 66 percent since 1968, according to the North American Breeding Bird Survey.
| Full article | 0 Comments | 20516 views |
| Bioinformatics | August 15, 2016 06:29 PM |

Manduca sexta caterpillars can grow up to 10 cm long making them ideal for laboratory experiments to investigate their biochemistry and physiology. "Whooo ... are ... you?" asked the hookah-smoking caterpillar of Alice, in Wonderland. Asking the question of the caterpillar instead, an international team of scientists have published their findings from the sequencing, annotation, and exploration of the genome of the tobacco hornworm moth. The project involved 114 scientists from 50 research institutions worldwide, including from the SIB Swiss Institute of Bioinformatics and the University of Geneva (UNIGE). This remarkable moth and strikingly beautiful and very large caterpillar are known by the Latin name, Manduca sexta, but also as the tobacco hornworm, Carolina sphinx moth, goliath worm, or 'Le sphinx du tabac'. The giant caterpillars are a favourite childhood pet, but they are considered serious agricultural pests as they feed voraciously on tobacco, potato, tomato, and pepper plants. The large size of the caterpillars means that this moth has become one of the most important model species for studying insect physiology, biochemistry, and molecular biology, and sequencing its genome opens many new research avenues.
| Full article | 0 Comments | 17158 views |
| Bioinformatics | July 27, 2016 05:24 PM |

This is a comparison of ancient rice remains and modern rice. Rice, or Oryza sativa as its scientifically known, feeds more than a third of the globe. Yet the majority of rice crops that supply 90 percent of the world come from just two domesticated varieties, japonica and indica.
| Full article | 0 Comments | 15415 views |
| Bioinformatics | July 25, 2016 06:09 PM |
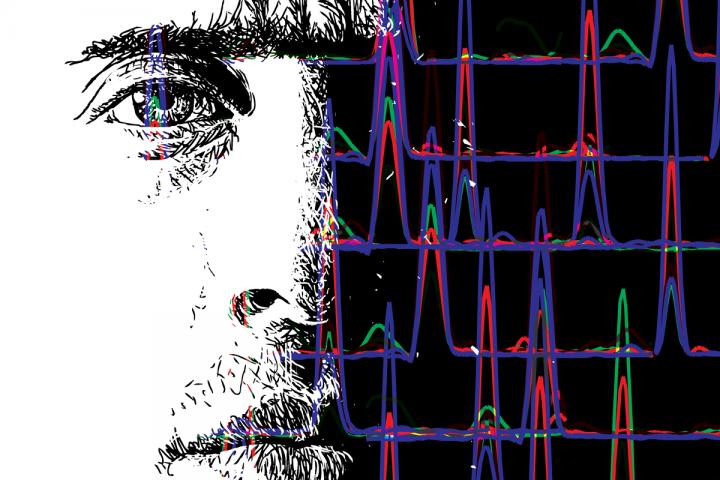
This is an illustration of SRM peaks and a human face. Reporting in the journal Cell, Senior Research Scientist Dr. Ulrike Kusebauch, of Institute for Systems Biology (ISB), describes the results of a collaboration between scientists at ISB, ETH Zurich and a number of other contributing institutes to develop the Human SRMAtlas, a compendium of proteomic assays for any human protein. The Human SRMAtlas is a compendium of highly specific mass spectrometry assays for the targeted identification and reproducible quantification of any protein in the predicted human proteome, including assays for many spliced variants, non-synonymous mutations and post-translational modifications. Using the technique called selected reaction monitoring, assays were developed with the use of 166,174 well-characterized, chemically synthesized proteotypic peptides. The SRMAtlas resource is freely publicly available at http://www.
| Full article | 0 Comments | 13950 views |
| Bioinformatics | July 18, 2016 06:59 PM |

Right: Photograph during excavation exhibiting excellent dry preservation of plant remains
Left: A well-preserved, desiccated barley grain found at Yoram Cave An international team of researchers has succeeded for the first time in sequencing the genome of Chalcolithic barley grains. This is the oldest plant genome to be reconstructed to date. The 6,000-year-old seeds were retrieved from Yoram Cave in the southern cliff of Masada fortress in the Judean Desert in Israel, close to the Dead Sea. Genetically, the prehistoric barley is very similar to present-day barley grown in the Southern Levant, supporting the existing hypothesis of barley domestication having occurred in the Upper Jordan Valley.
| Full article | 0 Comments | 14140 views |
| Bioinformatics | July 11, 2016 05:01 PM |
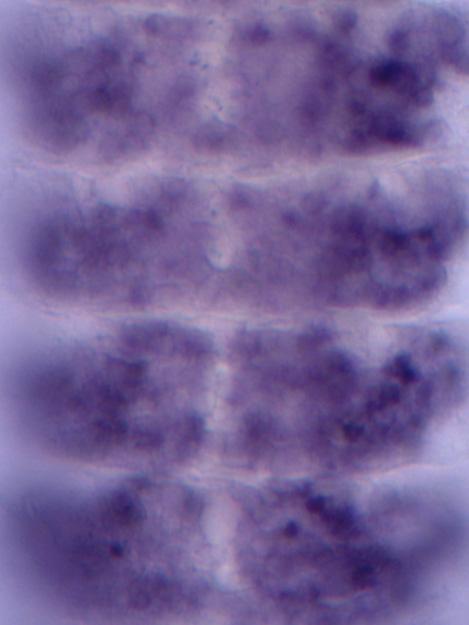
A microscope image shows a four-segment section of the nervous system of an Aedes aegypti mosquito embryo. The dark purple indicates areas where a gene called "short gastrulation " is being... Scientists are using machine learning to identify important sequences of DNA within the mosquito genome that regulate how the insect's cells develop and behave.
| Full article | 0 Comments | 14241 views |
| Bioinformatics | July 8, 2016 06:38 PM |
There are thousands of scientific papers dedicated to a particular type of tumor, a particular gene, a type of specific molecular lesion or the effect of a particular drug. However, there are very few examples of publications that integrate these four concepts (type of tumor, gene alteration and drug) in a significant amount of samples. An article published in Cell, in collaboration with the group of Dr. Manel Esteller, Director of the Epigenetics and Cancer Biology Program at the Bellvitge Biomedical Research Institute (IDIBELL), ICREA researcher and Professor of Genetics at the University of Barcelona, provides us with this important source of information.
| Full article | 0 Comments | 13591 views |
| Bioinformatics | June 29, 2016 04:50 PM |
Tumor DNA is cluttered with genomic alterations, the vast majority of which have little or no functional or clinical relevance. This means that even when cancer researchers discover an alteration in a tumor or a line of cancer cells, the alteration may or may not be relevant to the progression of the disease - chances are good (and history has shown) that many alterations that are correlated with cancer are not causative of cancer; many alterations are "passengers" rather than "drivers". A University of Colorado Cancer Center study published in the journal Cancer Research demonstrates a novel method for sorting passenger from driver alterations, and uses this method to pinpoint a new driver and potential therapeutic target in cancer progression, GON4L.
| Full article | 0 Comments | 13059 views |
| Bioinformatics | June 27, 2016 05:29 PM |
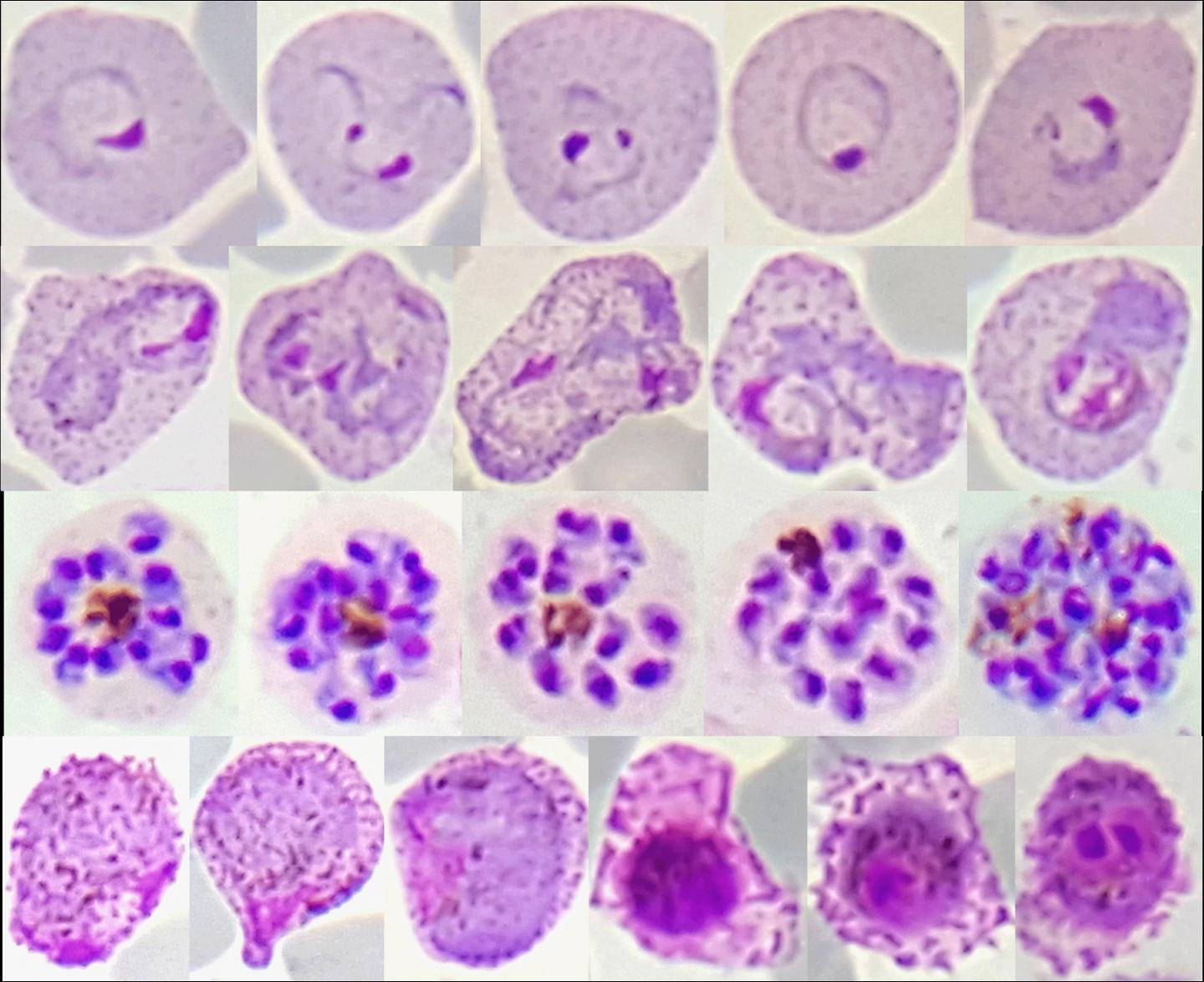
Red blood cell stages of Plasmodium vivax from malaria patients in Thailand. Plasmodium vivax (P. vivax) parasites, which cause a debilitating form of malaria, are yielding their secrets to an international team of researchers funded by the National Institute of Allergy and Infectious Diseases (NIAID), part of the National Institutes of Health. In the largest such effort to date, the team determined complete genomes of nearly 200 P. vivax strains that recently infected people in eight countries. Comparative analysis showed the parasites clustered into four genetically distinct populations that provide insights into the movement of P. vivax over time and suggest how it is still adapting to regional variations in both the mosquitoes that transmit it and the humans it infects.
| Full article | 0 Comments | 8231 views |
| Bioinformatics | June 21, 2016 03:52 PM |

CRG scientists of the Barcelona Metasub Team at the Global Sampling Day. Barcelona takes part in the international research project Metasub, which aims to map the microbiome of public transit systems in 54 cities worldwide, including New York, Hong Kong, Paris or Sydney.
| Full article | 0 Comments | 7436 views |
| Bioinformatics | May 25, 2016 05:44 PM |

This image shows a range of vertebrate the G10K members are working on: Bird - Ruby-throated Hummingbird; Reptile - Green Anole; Fish - Spotted Gar; Mammal - Koala The Genome Analysis Centre (TGAC) are to hold the biannual Genome 10K Conference on 29 August - 1 September 2017.
| Full article | 0 Comments | 6348 views |
| Bioinformatics | May 24, 2016 06:13 PM |
Whole-exome sequencing of both colorectal adenomas (precancers often called polyps) and intestinal mucosa at risk for developing into adenomas from patients with familial adenomatous polyposis (FAP) has generated a comprehensive picture of the genomic alterations that characterize the evolution of normal mucosa to precancer.
| Full article | 0 Comments | 5541 views |
| Bioinformatics | April 21, 2016 04:10 PM |
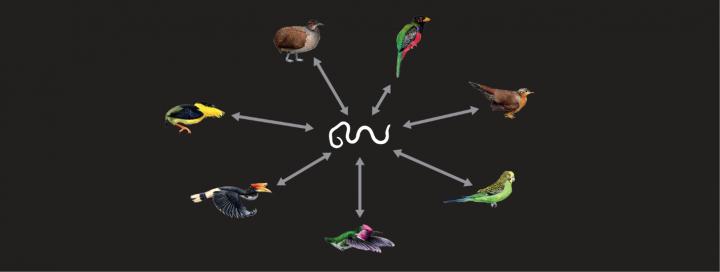
The researchers found DNA 'fossils' of parasitic nematodes in seven groups of birds (clockwise): trogons, mesites, parrots, hummingbirds, hornbills, manakins, tinamous. In rare instances, DNA is known to have jumped from one species to another. If a parasite's DNA jumps to its host's genome, it could leave evidence of that parasitic interaction that could be found millions of years later -- a DNA 'fossil' of sorts. An international research team led from Uppsala University has discovered a new type of so-called transposable element that occurred in the genomes of certain birds and nematodes.
| Full article | 0 Comments | 6542 views |
| Bioinformatics | April 18, 2016 06:13 PM |
Researchers at The University of Texas MD Anderson Cancer Center have announced a new method for detecting DNA mutations in a single cancer cell versus current technology that analyzes millions of cells which they believe could have important applications for cancer diagnosis and treatment. The results are published in the April 18 online issue of Nature Methods.
| Full article | 0 Comments | 6631 views |
| Bioinformatics | April 12, 2016 04:22 PM |

Queen and worker ants develop from the same sets of genes, but end up being structurally, behaviourally, and functionally different. Queen and worker ants develop from the same sets of genes, but perform completely different ecological roles. How the same genes result in two types of individuals is an ongoing mystery. In the past, scientists have only studied a small number of ant species at a time to try to understand the nature of queen-worker differences. However, a team from the Okinawa Institute of Science and Technology Graduate University (OIST) in tandem with the University of Helsinki and other collaborators from around the world, recently looked at a large data set with 16 species that provides insight into the differences between queen and worker ants.
| Full article | 0 Comments | 7846 views |
| Bioinformatics | March 22, 2016 06:46 PM |

Female orphan chimpanzee are cared for at the Sanaga-Yong Chimpanzee Rescue Center. Understanding the origins of emerging diseases - as well as more established disease agents -- is critical to gauge future human infection risks and find new treatment and prevention approaches. This holds true for malaria, which kills more than 500,000 people a year. Symptoms, including severe anemia, pregnancy-associated malaria, and cerebral malaria, have been linked to the parasite's ability to cause infected red blood cells to bind to the inner lining of blood vessels.
| Full article | 0 Comments | 7492 views |
| Bioinformatics | March 14, 2016 06:16 PM |

This image shows Yucca Brevifolia Tikaboo, June 2014. Scientists at the Donald Danforth Plant Science Center have teamed up with researchers at Willamette University, a liberal arts college in Salem Oregon, to develop genetic tools that could save the Joshua tree from extinction. Together with scientists from The University of Georgia and the University of British Columbia, and with the support of several Mojave Desert conservation organizations, researchers are inviting members of the public to help get the project off the ground by making donations at the crowdfunding site Experiment.com. In the past two weeks, more than 100 backers have donated more than $4,000 to The Joshua Tree Genome Project. The project aims to raise $8,500 by March 24th.
| Full article | 0 Comments | 6537 views |
| Bioinformatics | March 9, 2016 05:04 PM |
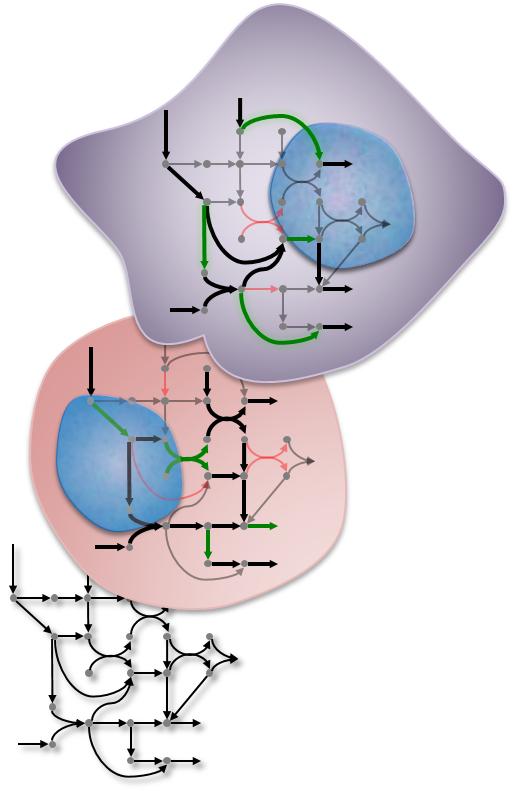
The same metabolic pathway can produce different results in different bodily tissues. A Rice University algorithm is designed to find those differences. Rice University bioengineers have introduced a fast computational method to model tissue-specific metabolic pathways. Their algorithm may help researchers find new therapeutic targets for cancer and other diseases.
| Full article | 0 Comments | 6366 views |
| Bioinformatics | March 9, 2016 05:04 PM |
Led by Drs John Postlethwait and Ingo Braasch from the Institute of Neuroscience, University of Oregon, US, in collaboration with the Broad Institute, the study of the Spotted Gar (Lepisosteus oculatus) genome reveals that it is small and manageable. Furthermore, it lacks much shuffling and duplication that occurred in the 'main' fish ancestral line; it conserved its genome.
| Full article | 0 Comments | 6474 views |
| Bioinformatics | February 16, 2016 05:20 PM |

These bloodsuckers originally parasitized bats. The bed bug (Cimex lectularius) has been a familiar human parasite for more than 3,000 years. After a significant decrease in its population density in the middle of the last century, we have seen a dramatic increase again around the world over the past 20 years. In Australia, for instance, there is an increase of 4,500%.
| Full article | 0 Comments | 8944 views |
| Bioinformatics | February 9, 2016 07:45 PM |

Researchers sequence the genome of the Lyme-disease-causing tick and find lots of duplicative elements. Researchers have sequenced the genetic blueprint of one of the most prolific pathogen-transmitting agents on the planet - the Lyme-disease-spreading tick (Ixodes scapularis) that bites humans. The findings could lead to advances in not only disrupting the tick's capacity to spread diseases but also in eradicating the pest.
| Full article | 0 Comments | 7532 views |
| Bioinformatics | February 8, 2016 04:12 PM |
Database searches for DNA sequences that can take biologists and medical researchers days can now be completed in a matter of minutes, thanks to a new search method developed by computer scientists at Carnegie Mellon University.
| Full article | 0 Comments | 8384 views |
| Bioinformatics | January 19, 2016 03:57 PM |
A team of scientists from Germany, USA, and Russia, including Dr. Mark Borodovsky, a Chair of the Department of Bioinformatics at MIPT, have proposed an algorithm to automate the process of searching for genes, making it more efficient. The new development combines the advantages of the most advanced tools for working with genomic data. The new method will enable scientists to analyse DNA sequences faster and more accurately and identify the full set of genes in a genome.
| Full article | 0 Comments | 7278 views |
| Bioinformatics | January 6, 2016 07:55 PM |
In October, an interdisciplinary group of scientists proposed forming a Unified Microbiome Initiative (UMI) to explore the world of microorganisms that are central to life on Earth and yet largely remain a mystery. An article in the journal ACS Nano describes the tools scientists will need to understand how microbes interact with each other and with us.
| Full article | 0 Comments | 6676 views |
| Bioinformatics | December 28, 2015 06:40 PM |
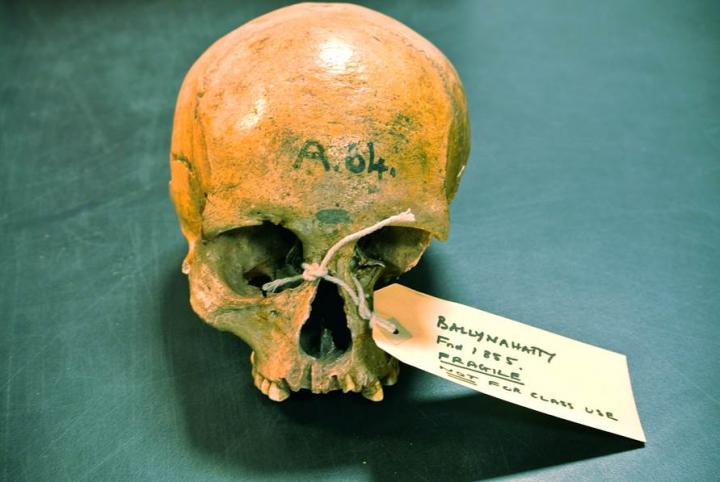
Excavated near Belfast in 1855, she had lain in a Neolithic tomb chamber for 5,000 years; subsequently curated in Queens University Belfast. A team of geneticists from Trinity College Dublin and archaeologists from Queen's University Belfast has sequenced the first genomes from ancient Irish humans, and the information buried within is already answering pivotal questions about the origins of Ireland's people and their culture.
| Full article | 0 Comments | 9724 views |
| Bioinformatics | December 23, 2015 04:13 AM |
Researchers long have known that some portion of the risk of developing cancer is hereditary and that inherited genetic errors are very important in some tumors but much less so in others.
| Full article | 0 Comments | 9626 views |
| Bioinformatics | December 9, 2015 06:53 PM |
Oncologists are increasingly using information obtained from investigations of the tumor genome in order to find individualized therapies for patients. They specifically search the hereditary information of cancer cells for mutations that drive malignant growth. By now, targeted drugs against many of these cancer-typical cellular alterations have become available.
| Full article | 0 Comments | 11412 views |
| Bioinformatics | December 9, 2015 06:53 PM |
Scientists have performed the first comprehensive genomic analysis of Ebola virus sequences from Liberia, one of three countries widely affected by the devastating outbreak that began in 2013 in Western Africa. Their work, published today in Cell Host & Microbe, traces the introduction and spread of the virus in Liberia and also sheds light on how the virus moved between the neighboring countries of Guinea and Sierra Leone.
| Full article | 0 Comments | 9770 views |
Return to Biology News Net Homepage
- In some genetic cases of microcephaly, stem cells fail to launch
- Yale team discovers how Zika virus causes fetal brain damage
- Scientists uncover the way a common cell enzyme alerts the body to invading bacteria
- Arctic gives clues on worst mass extinction of life
- Calcium channel blockers caught in the act at atomic level
- Flexibility That A.C.A. Lent to Work Force Is Threatened by...International Herald Tribune
- Bacteria Behind Legionnaires’ Disease Found at New York Police StationInternational Herald Tribune
- Prescription Price Crisis? 28 Million Americans See Spike in Drug...MSNBC.com: Health
- Seizing on Opioid Crisis, a Drug Maker Lobbies Hard for...International Herald Tribune
- Republicans are taking a big political risk on health careAP Health News
- Why We're So Divided Over Saving WolvesNational Geographic News
Top Biology News about Bioinformatics
Biology Articles & News
Last 7 Days
Biology Articles & News
Most popular ever in Bioinformatics
- Giraffes more speciose than expected (496403)
- Artificial intelligence expedites breast cancer risk prediction (64537)
- Touching Molecules With Your Bare Hands (23134)
- After the first decade of metagenomics -- adolescent growth spurt anticipated (21817)
- NHGRI announces latest sequencing targets (21318)
- Computer-assisted drug design for Malaria (20604)
- Warbler genomes look to be 99.97 percent alike (20516)